Vektorer är DNA-molekyler (t.ex. plasmider) som har specifika klipp- och infogningsställen där man kan sätta in gener för kloning eller uttryck.
När man klipper DNA använder man restriktionsenzymer, som känner igen och klyver specifika sekvenser.
MCS (multiple cloning site) är en kort DNA-region i en vektor som innehåller många unika restriktionsställen där man kan klippa och sätta in gener.
Man ligerar DNA med DNA-ligas, ett enzym som kopplar ihop ändarna genom att skapa fosfodiesterbindningar.
BamH I är ett restriktionsenzym
Klyvning kan ge två typer av ändar: sticky ends (överhängande ändar) och blunt ends (trubbiga ändar).
E. coli används eftersom den växer snabbt, är lätt att odla och genetiskt manipulera, och vi känner dess genom mycket väl.
Dag 1 & 2:
- Genen lacZ i E. coli kodar för enzymet β-galaktosidas
- IPTG är en inducer som behövs för att slå på lacZ, så att enzymet faktiskt bildas
- X-gal är ett konstgjort substrat, som blir blått när de klyvs av β-galaktosidas
- X-gal och 5-Bromo-4-Chloro-3-Indolyl β-D-Galactopyranoside är samma sak
- Normalt klyver enzymet β-galaktosidas X-gal → blått färgämne bildas
- För att substratet ska bli blått krävs lacZ, IPTG och X-gal. Saknas några av dem blir det inte blått
- Ampicillin dödar bakterier som inte har plasmiden, så bara de vi vill studera överlever.
- Heat-shock 0→37 grader skapar en tryckskillnad och öppnar porer i membrandet så plasmid-DNA kan ta sig in i cytoplasman
- Nerkylning direkt efteråt gör att de stannar kvar
- LB-plattan är både odlingsmedium och selektions-/screeningsverktyg
- näring (aminosyror, mineral, salt, tillväxtfaktorer osv)
- ampicillin dödar allt utan plasmid
- IPTG slår på lacZ
- X-gal infärgning
Dag 3
A. Förbered plasmiden
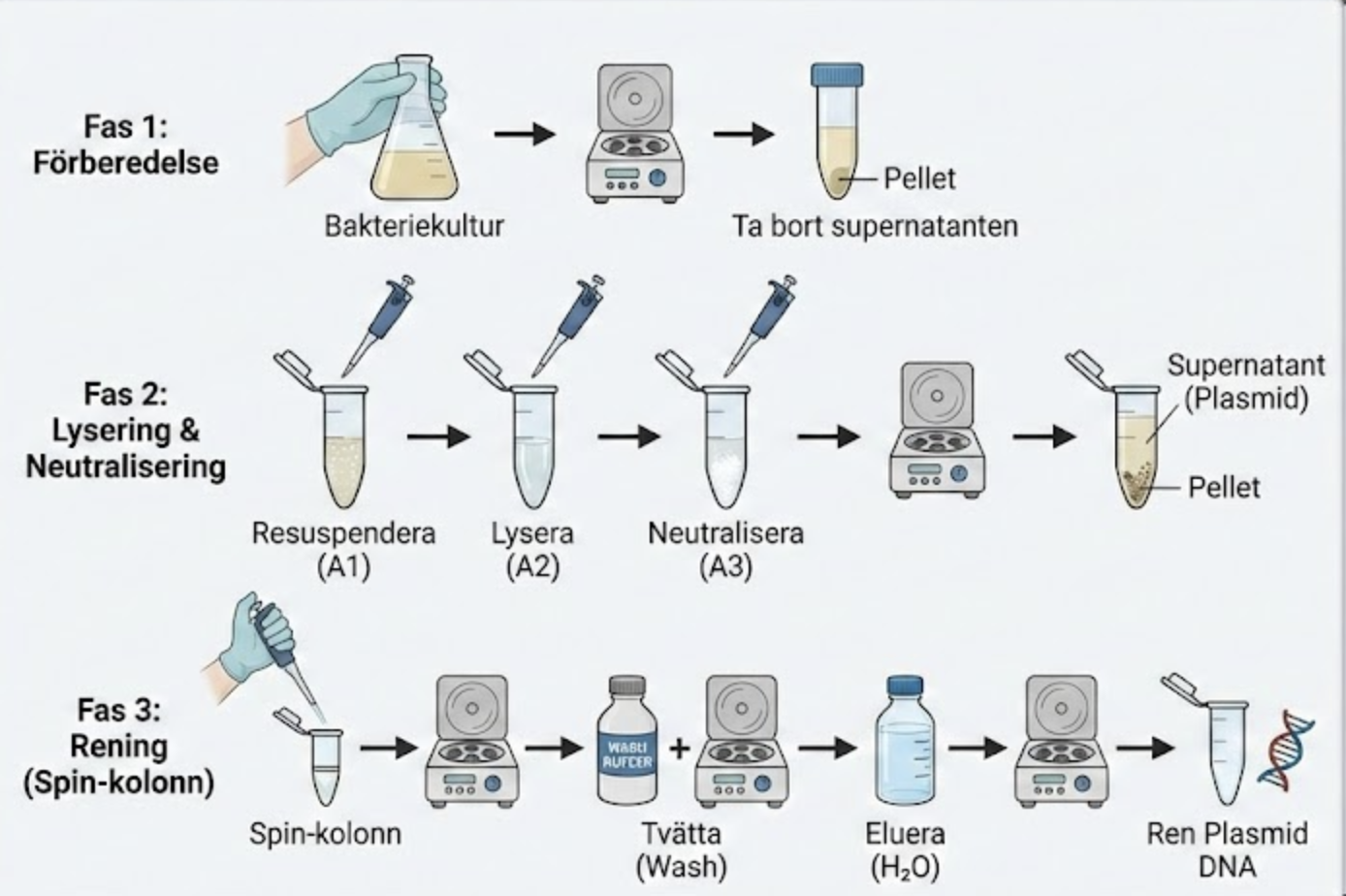
Cellerna centrifugeras, spräcks och plasmiden separeras från cellskräpet. Plasmiden fångas sedan upp i en spin-kolonn, tvättas ren och sköljs ut i en ny tub.
Vi har två lösningar i separata falkonrör, en blå (utan våran insert) och en vit (med vår insert). Upprepa alla stegen för varje lösning.
| Steg | Input | Output | Kort beskrivning |
|---|---|---|---|
| 1. Pelletera | Bakteriekultur | Cellpellet + borttagen supernatant | Celler centrifugeras ned till botten. |
| 2. Lös upp | Cellpellet + A1 | Jämn cellsuspension | Löser upp pelleten så lysering kan fungera. |
| 3. Lysera | Cellsuspension + A2 | Lyserad lösning | Cellmembran öppnas, plasmid + DNA/proteiner frigörs. |
| 4. Neutralisera | Lyserad lösning + A3 | Fällt skräp + plasmid i lösn. | Stora DNA/proteiner klumpar ihop sig. Plasmiden stannar löslig. |
| 5. Centrifugera | Neutraliserad mix | Pellet (skräp) + supernatant (plasmid) | Plasmiden separeras från cellskräp. |
| 6. Ladda kolumn | Supernatant | Plasmid bundet till kolumn | Plasmiden fastnar på membranet; skräp rinner igenom. |
| 7. Tvätta | Kolumn + A4 | Renad kolumn | Tar bort salter, proteiner och kvarvarande föroreningar. |
| 8. Torka kolumn | Tvättad kolumn | Torr kolumn | Extra spinn för att avlägsna etanolrester. |
| 9. Skölj | Kolumn + vatten | Ren plasmid-DNA i eppendorf | Vattnet löser av plasmiden från membranet. |
| Vi får nu ut en ren plasmid |
B. klyv plasmiden med BamHI och jämför den med en oklippt kontroll.
Skapa en BamHI-enzym som kan klippa plasmiden, klipp plasmiden och tillsätt färgnings
Master solution:
- vatten, buffert och enzym blandas in
Digested = cut = plasmid som har blivit klippt vid BamHI-sajten Cybr safe gör det synligt i UV-ljus utan att vara cancerframkallande loading dye gör att vi kan se vad vi pipeterar in i elektroforesen i nästa steg
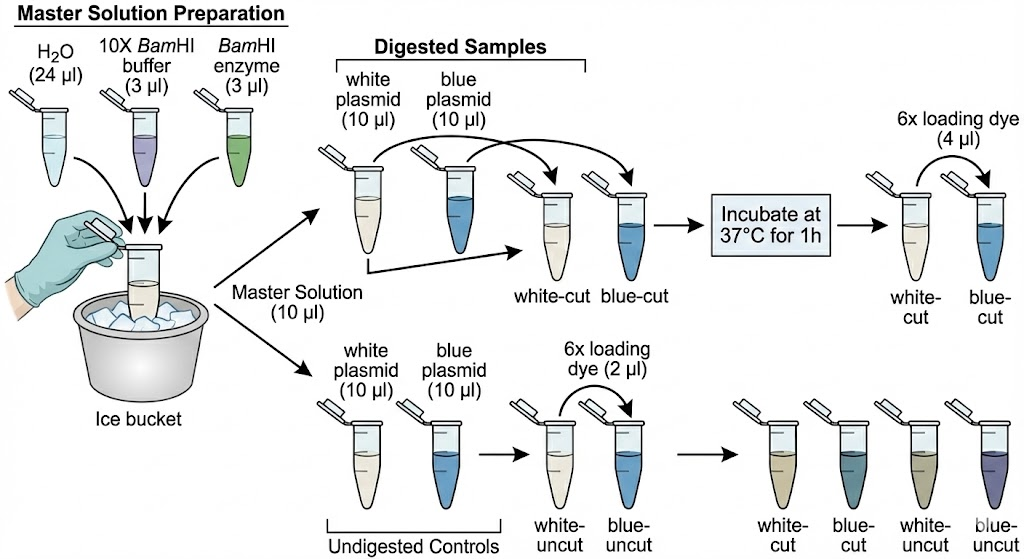
| Steg | Input | Output | Kort beskrivning |
|---|---|---|---|
| 1. Gör BamHI-mastermix | H₂O + 10× buffer + BamHI | Färdig enzymmix | Blandar så alla prover får samma enzym och buffer. |
| 2. Fördela plasmider | Plasmid-DNA | Tub “white-cut” + tub “blue-cut” | Flyttar plasmiderna till nya rör för klippning. |
| 3. Tillsätt master + inkubera | Plasmid + BamHI-mastermix | Digesterat (klippt) DNA | Enzymet klipper plasmiden vid BamHI-sajten. |
| 4. Tillsätt loading dye | Klippt DNA + dye | Färdiga “cut”-prover för gel | Gör proverna tyngre och synliga i gelen. |
| 5. Gör oklippta kontroller | Plasmid + loading dye | “white-uncut” + “blue-uncut” | Referensprover som visar hur odigesterat DNA ser ut. |
Du blandar en BamHI-lösning, blandar den med plasmiderna och låter enzymet klippa DNA:t vid 37 °C. Sedan tillsätter du loading dye och gör även två oklippta kontroller för jämförelse i gelen.
Steg 3
Elektrofoera och läs av i UV-ljus
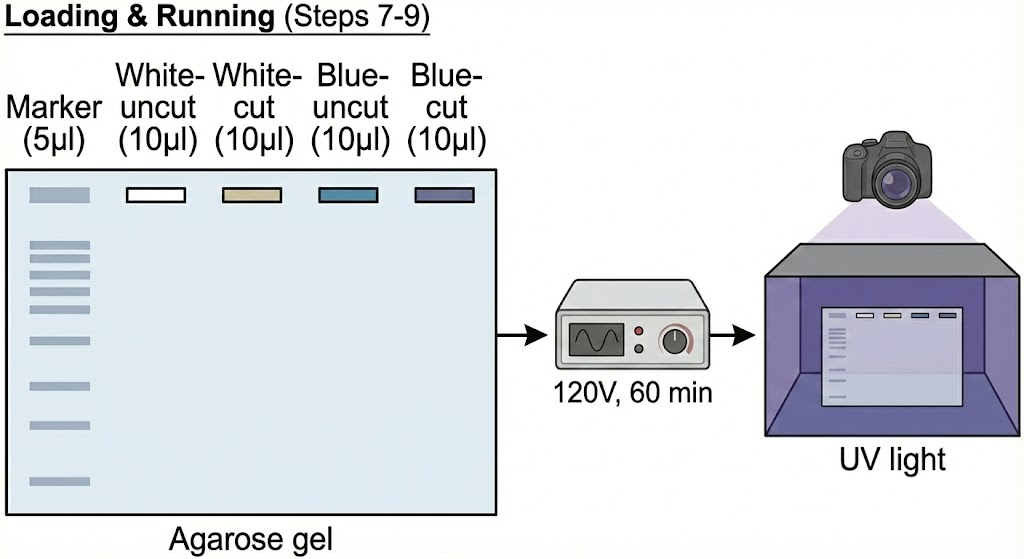
| Steg | Input | Output | Kort beskrivning |
|---|---|---|---|
| 7. Ladda prover | Marker + cut/uncut-prover | Gel med laddade prover | Proverna placeras i rätt ordning i brunnarna. |
| 8. Kör elektrofores | Gel i tank + 120 V | Separering av DNA-fragment | DNA vandrar efter storlek genom gelen. |
| 9. Fotografera | Färdigkörd gel | UV-bild med DNA-band | Bandmönstret visar plasmidens storlek/insätt. |